Kromosoomid
Definitsioon - mis on kromosoomid?
Raku geneetiline koostis on salvestatud DNA (deoksüribonukleiinhape) ja selle aluste (adeniin, tümiin, guaniin ja tsütosiin) kujul. Kõigis eukarüootsetes rakkudes (loomad, taimed, seened) on see rakutuumas kromosoomide kujul. Kromosoom koosneb ühest koherentsest DNA molekulist, mis on seotud teatud valkudega.
Kromosoomi nimi on tuletatud kreeka keelest ja seda saab umbkaudu tõlkida kui "värvkeha". See nimi tuleneb asjaolust, et väga varajases tsütoloogiaajaloos (1888) õnnestus teadlastel need spetsiaalsete põhivärvide abil värvida ja valgusmikroskoobis tuvastada. Kuid need on tegelikult nähtavad ainult rakutsükli teatud punktis, mitoos (sugurakkudes, meioos), kui kromosoom on eriti tihe (kondenseerunud).

Kuidas ehitatakse kromosoome?
Kui kogu raku DNA kahekordne spiraal, s.o umbes 3,4 x 109 aluspaari, ühendataks, oleks selle pikkuseks üle ühe meetri. Kõigi lisatud kromosoomide kogupikkus on ainult umbes 115 um. Seda pikkuse erinevust seletatakse kromosoomide väga kompaktse struktuuriga, milles DNA mitu korda väga spetsiifiliselt keritakse või keeratakse.
Histoonid, valkude erivorm, mängivad selles olulist rolli. Kokku on 5 erinevat histooni: H1, H2A, H2B, H3 ja H4. Viimasest neljast histoonist kaks moodustavad silindrikujulise struktuuri - oktameeri -, mille ümber kahekordne spiraal keerleb umbes kaks korda (= superheeliks). H1 kinnitub selle struktuuri külge, et seda stabiliseerida.
Seda DNA, oktameeri ja H1 kompleksi nimetatakse nukleosoomiks. Mitmed neist nukleosoomidest on nüüd suhteliselt lühikese intervalliga (10–60 aluspaari) üksteise järel „nagu pärlijoon”. Kromosoomide vahelisi lõike nimetatakse speisserver-DNA-ks. Üksikud nukleosoomid puutuvad nüüd uuesti kokku H1 kaudu, mis loob täiendava spiraali ja seega ka kompressiooni.
Saadud ahel on omakorda silmustes, mida stabiliseerib happelistest mitteh histoonvalkudest koosnev selgroog, tuntud ka kui hertoonid. Need silmused esinevad omakorda valkude poolt stabiliseeritud spiraalides, mille tulemuseks on kokkusurumise viimane etapp. Kuid see suur kokkusurumisaste ilmneb ainult raku jagunemise kontekstis mitoosi ajal.
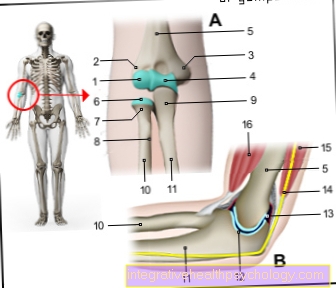
Selles faasis näete ka kromosoomide iseloomulikku kuju, mis koosneb kahest kromatiidist. Nende ühenduskohta nimetatakse tsentromeeriks. See jagab iga metafaasilise kromosoomi kaheks lühikeseks ja kaheks pikaks käsivarreks, mida nimetatakse ka p ja q käsivarreks.
Kui tsentromeer asub umbes kromosoomi keskel, nimetatakse seda metatsentriliseks kromosoomiks, kui see asub täielikult akrotsentrilise kromosoomi ühes otsas. Vahepealseid nimetatakse submetatsentrilisteks kromosoomideks. Need erinevused, mida on juba valguse mikroskoobi all näha, koos pikkusega võimaldavad kromosoomid esialgu klassifitseerida.
Mis on telomeerid?
Telomeerid on korduvate järjestustega kromosoomide otsad (TTAGGG). Need ei sisalda mingit asjakohast teavet, vaid pigem aitavad vältida asjakohasemate DNA lõikude kadumist. Iga raku jagunemisega kaob osa kromosoomist DNA replikatsiooni mehhanismi kaudu.
Niisiis on telomeerid teatud mõttes puhvrid, mis viivitavad punktiga, kus rakk jagades jagab olulist teavet. Kui raku telomeeride pikkus on alla 4000 aluspaari, alustatakse programmeeritud rakusurma (apoptoos). See hoiab ära vigase geneetilise materjali leviku organismis. Mõnel rakul on telomeraasid, st ensüümid, mis on võimelised telomeere uuesti pikendama.
Lisaks tüvirakkudele, millest pärinevad kõik teised rakud, on need sugurakud ja teatud immuunsüsteemi rakud. Lisaks leidub vähirakkudes ka telomeraase, mistõttu räägitakse selles kontekstis raku surematustamisest.
Loe kõike selle teema kohta siit: Telomeerid - anatoomia, funktsioon ja haigused
Mis on kromatiin?
Kromatiin viitab kogu rakutuuma sisule, mida saab alusega värvida. Seetõttu hõlmab termin lisaks DNA-le ka teatud valke, näiteks histoone ja hertoone (vt struktuur), samuti teatud RNA fragmente (hn ja snRNA).
Sõltuvalt rakutsükli faasist või geneetilisest aktiivsusest on see materjal saadaval erineva tihedusega. Tihedamat vormi nimetatakse heterokromatiiniks. Mõistmise hõlbustamiseks võiks seetõttu pidada seda "säilitamisvormiks" ja teha siin jälle vahet konstitutiivsel ja fakultatiivsel heterokromatiinil.
Konstitutiivne heterokromatiin on kõige tihedam vorm, mis esineb rakutsükli kõigis faasides kõige kõrgemal kondenseerumisel. See moodustab umbes 6,5% inimese genoomist ja asub peamiselt vähesel määral tsentromeeride ja kromosoomiharude (telomeeride) otste lähedal, aga ka muudes kohtades (peamiselt kromosoomid 1, 9, 16, 19 ja Y) . Lisaks paikneb suurem osa konstitutiivsest heterokromatiinist tuumamembraani lähedal, see tähendab rakutuuma servades. Keskel asuv ruum on reserveeritud aktiivsele kromatiinile, eukromatiinile.
Fakultatiivne heterokromatiin on veidi vähem tihe ja seda saab vastavalt vajadusele või sõltuvalt arenguastmest aktiveerida ja deaktiveerida. Hea näide selle kohta on naiste karüotüüpide teine X-kromosoom. Kuna raku ellujäämiseks piisab põhimõtteliselt ühest X-kromosoomist, nagu meestele lõpuks piisab, deaktiveeritakse embrüonaalses faasis üks neist kahest. deaktiveeritud X-kromosoom on tuntud kui Barri keha.
Ainult rakkude jagunemise ajal kondenseerub see mitoosi kontekstis täielikult, saavutades metafaasis kõrgeima kokkusurumise. Kuna aga erinevaid geene loetakse sageli erinevalt - lõppude lõpuks ei nõuta kõiki valke kogu aeg ühesuguses koguses -, siis eristatakse siin ka aktiivset ja passiivset eukromatiini.
Lisateavet selle kohta leiate: Kromatiin
Haploidsed kromosoomid
Haploid (kreeka keeles haploos = üksik) tähendab, et kõik raku kromosoomid esinevad eraldi, s.t mitte paarides (diploidsed), nagu tavaliselt juhtub. See on kõigi munarakkude ja seemnerakkude loomulik seisund, kus kaks identset kromatiidi ei eraldata esimese meioosi käigus esialgu, vaid kõigepealt eraldatakse kõik kromosoomipaarid.
Selle tulemusena on pärast esimest meioosi inimestel tütarrakkudel tavapärase 46 kromosoomi asemel ainult 23, mis vastab poolele haploidsele kromosoomikomplektile. Kuna nendel tütarrakkudel on endiselt iga 2 kromosoomist koosneva kromosoomi identne koopia, on vajalik teine meioos, milles kaks kromatiidi on üksteisest eraldatud.
Polüteenikromosoomid
Polüteenkromosoom on kromosoom, mis koosneb suurest hulgast geneetiliselt identsetest kromatiididest. Kuna selliseid kromosoome on isegi väiksema suurenduse korral lihtne näha, nimetatakse neid mõnikord hiidkromosoomideks. Selle eelduseks on endoreplikatsioon, mille käigus rakutuumas paiknevad kromosoomid korrutatakse mitu korda ilma rakujagunemiseta.
Millised on kromosoomide funktsioonid?
Kromosoom kui meie genoomi organisatsiooniline üksus aitab peamiselt tagada, et kahekordistunud genoom jaotuks rakkude jagunemise ajal tütarrakkude vahel ühtlaselt. Selleks tasub lähemalt uurida rakkude jagunemise mehhanisme või rakutsüklit:
Rakk veedab suurema osa rakutsüklist interfaasis, mis tähendab kogu ajaperioodi, mille jooksul rakk ei hakka kohe jagunema. See jaguneb omakorda faasideks G1, S ja G2.
G1 faas (G nagu lõhe, st lõhe) järgneb kohe rakkude jagunemisele. Siin suureneb raku suurus uuesti ja täidab üldisi metaboolseid funktsioone.
Siit saab see üle minna ka G0 faasile. See tähendab, et see muutub etapiks, mis ei ole enam jagunemisvõimeline, ja tavapärastel juhtudel muutub see ka väga konkreetseks funktsiooniks (rakkude diferentseerumine). Nende ülesannete täitmiseks loetakse intensiivsemalt väga spetsiifilisi geene, teisi vähem või üldse mitte.
Kui DNA segmenti pole pikka aega vaja, asub see sageli kromosoomide osades, mis on pikka aega tihedalt pakitud (vt kromatiin). Ühelt poolt on selle eesmärk säästa ruumi, kuid lisaks muudele geeniregulatsiooni mehhanismidele on see ka lisakaitse juhusliku lugemise vastu. Siiski on täheldatud ka seda, et väga spetsiifilistes tingimustes võivad diferentseerunud rakud G0 faasist uuesti tsüklisse siseneda.
G1 faasile järgneb S faas, see tähendab faas, milles sünteesitakse uus DNA (DNA replikatsioon). Siin peab kogu DNA olema kõige lõdvemal kujul, see tähendab, et kõik kromosoomid on täielikult kerimata (vt struktuuri).
Sünteesifaasi lõpus on kogu geneetiline materjal rakus kahes eksemplaris. Kuna koopia on endiselt tsentromere kaudu kinnitatud algsele kromosoomile (vt struktuuri), ei räägita kromosoomide dubleerimisest.
Iga kromosoom koosneb nüüd ühe asemel kahest kromatiidist, et see saaks hiljem mitoosi ajal omandada iseloomuliku X-kuju (rangelt öeldes kehtib X-kuju ainult metatsentriliste kromosoomide kohta). Järgmises G2 faasis toimub kohene ettevalmistus rakkude jagunemiseks. See hõlmab ka replikatsioonivigade ja ahelate purunemiste üksikasjalikku kontrollimist, mida saab vajadusel parandada.
Rakkude jagunemist on põhimõtteliselt kahte tüüpi: mitoos ja meioos. Välja arvatud sugurakud, tekivad mitoosi kaudu kõik organismi rakud, mille ainus ülesanne on kahe geneetiliselt identse tütarraku moodustamine.
Meioosi eesmärk on seevastu genereerida geneetiliselt erinevaid rakke:
Esimeses etapis jagatakse vastavad (homoloogsed), kuid mitte identsed kromosoomid. Alles järgmises etapis eraldatakse kromosoomid, mis koosnevad kahest identsest kromatiidist ja jaotatakse uuesti mõlemale kahele tütarrakule, nii et lõpuks tekivad ühest eellasrakust neli erineva geneetilise materjaliga sugurakku.
Kromosoomide vorm ja struktuur on mõlema mehhanismi jaoks hädavajalikud: Spetsiaalsed "valgulõngad", nn spindli aparaadid, kinnituvad tugevalt kondenseerunud kromosoomide külge ja tõmbavad kromosoomid peenelt reguleeritud protsessis kesktasandist (ekvatoriaaltasandist). ühtlase jaotuse tagamiseks ühe ümber oleva raku vastupoolustele. Isegi väikestel muutustel kromosoomide mikrostruktuuris võivad siin olla tõsised tagajärjed.
Kõigil imetajatel määrab sugukromosoomide X ja Y suhe ka järglaste soo. Põhimõtteliselt sõltub kõik sellest, kas munarakuga ühineval spermal on X- või Y-kromosoom. Kuna mõlemat sperma vormi toodetakse alati täpselt samas ulatuses, on tõenäosus mõlema sugupoole jaoks alati tasakaalus. See juhuslik süsteem tagab soolise jaotuse ühtlasema kui näiteks keskkonnategurite, näiteks temperatuuri puhul.
Lisateave teema kohta: Rakutuuma jagunemine
Kuidas edastatakse geneetiline koostis kromosoomide kaudu?
Täna teame, et tunnused on pärilikud geenide kaudu, mis on rakkudesse talletatud DNA kujul. Need jagunevad omakorda 46 kromosoomiks, millel jaotub inimese 25 000–3 300 geeni.
Lisaks omadusele endale, mida nimetatakse fenotüübiks, on olemas ka geneetiline ekvivalent, mida nimetatakse genotüübiks. Geeni paiknemist kromosoomis nimetatakse lookuseks. Kuna inimestel on igas kromosoomis topelt, siis esineb iga geen ka kaks korda. Ainsaks erandiks on meeste X-kromosomaalsed geenid, kuna Y-kromosoom kannab ainult murdosa X-kromosoomis leiduvast geneetilisest teabest.
Erinevaid geene, mis asuvad samal lookusel, nimetatakse alleelideks. Sageli on ühes lookuses rohkem kui kaks erinevat alleeli. Seejärel räägitakse polümorfismist. Selline alleel võib olla lihtsalt kahjutu variant (normaalne variant), aga ka patoloogilised mutatsioonid, mis võivad olla päriliku haiguse käivitajad.
Kui fenotüübi muutmiseks piisab ühe geeni mutatsioonist, räägitakse monogeensest või Mendeli pärandist. Kuid paljud päritavad tunnused on päritud mitme vastastikmõjuva geeni kaudu ja seetõttu on neid palju raskem uurida.
Kuna ema ja isa annavad mõlemad Mendeli pärandis lapsele edasi ühe oma kahest geenist, on järgmises põlvkonnas alati neli võimalikku kombinatsiooni, kusjuures ka need võivad ühe omaduse suhtes olla samad. Kui indiviidi mõlemal alleelil on fenotüübile sama mõju, on indiviid selle tunnuse suhtes homosügootne ja tunnus on vastavalt täielikult väljendatud.
Heterosügootidel on kaks erinevat alleeli, mis saavad üksteisega erineval viisil suhelda: kui üks alleel on teise suhtes domineeriv, pärsib see selle ekspressiooni täielikult ja domineeriv omadus muutub fenotüübis nähtavaks. Mahasurutud alleeli nimetatakse retsessiivseks.
Koodominantse pärandi korral võivad mõlemad alleelid end väljendada üksteise poolt mõjutamata, vahepealse pärandi korral on aga segu mõlemast tunnusest. Hea näide selle kohta on AB0 veregrupisüsteem, kus A ja B on omavahel domineerivad, kuid 0 domineerivad üksteise üle.
Milline on inimeste normaalne kromosoomide komplekt?
Inimrakkudel on 22 soost sõltumatut kromosoomipaari (autosoomid) ja kaks sugukromosoomi (gonosoomid), seega kokku 46 kromosoomi moodustavad ühe kromosoomikomplekti.
Autosoomid tulevad tavaliselt paarikaupa. Paari kromosoomid on geenide kuju ja järjestuse poolest sarnased ning seetõttu nimetatakse neid homoloogseteks. Naiste kaks X-kromosoomi on samuti homoloogsed, samas kui meestel on X- ja Y-kromosoom. Need erinevad esinevate geenide vormi ja arvu poolest nii, et homoloogiast ei saa enam rääkida.
Sugurakkudel, s.o munarakkudel ja seemnerakkudel, on meioosi tõttu ainult pool kromosoomist, nimelt 22 üksikut autosoomi ja üks gonosoom. Kuna sugurakud viljastumisel sulanduvad ja vahetavad mõnikord terveid segmente (ristumine), luuakse uus kromosoomide kombinatsioon (rekombinatsioon). Kõiki kromosoome koos nimetatakse karüotüübiks, mis on väheste eranditega (vt kromosoomide kõrvalekalded) kõigil samasoolistel isikutel identne.
Siit saate teada kõike teema kohta: Mitoos - lihtsalt seletatav!
Miks on alati kromosoomipaare?
Põhimõtteliselt saab sellele küsimusele vastata ühe lausega: Sest see on osutunud kasulikuks.Kromosoomipaaride olemasolu ja rekombinatsiooni põhimõte on seksuaalse paljunemise seisukohalt pärilikkuse jaoks hädavajalikud. Nii võib kahe inimese geneetilisest materjalist juhuslikult välja tulla täiesti uus isend.
See süsteem suurendab tohutult liikide omaduste mitmekesisust ja tagab selle, et see suudab muutunud keskkonnatingimustega kohaneda palju kiiremini ja paindlikumalt, kui see oleks võimalik ainult mutatsiooni ja valiku abil.
Ka kahekordsel kromosoomikomplektil on kaitsev toime: kui geeni mutatsioon tooks kaasa funktsiooni tõrke, on teises kromosoomis ikkagi mingi "varukoopia". Alati ei piisa organismist talitlushäire kompenseerimiseks, eriti kui muteerunud alleel on domineeriv, kuid see suurendab selle tõenäosust. Lisaks sellele ei kandu mutatsioon automaatselt kõigile järglastele, mis omakorda kaitseb liiki liiga radikaalsete mutatsioonide eest.
Mis on kromosoomimutatsioon?
Geneetilised defektid võivad tekkida ioniseerivast kiirgusest (nt röntgenikiirgus), keemilistest ainetest (näiteks bensopüreen sigaretisuitsus), teatud viirustest (nt HP viirused) või väikese tõenäosusega võivad need tekkida ka täiesti juhuslikult. Selle väljatöötamisel on sageli mitu tegurit. Põhimõtteliselt võivad sellised muutused esineda kõigis kehakudedes, kuid praktilistel põhjustel piirdub analüüs tavaliselt lümfotsüütidega (spetsiaalne immuunrakkude tüüp), fibroblastidega (sidekoerakud) ja luuüdi rakkudega.
Kromosoomimutatsioon on üksikute kromosoomide peamine struktuurimuutus. Tervete kromosoomide puudumine või lisamine oleks seevastu genoomi või ploidia mutatsioon, samas kui termin geenimutatsioon viitab suhteliselt väikestele muutustele geenis. Mõiste kromosoomide aberratsioon (ladina keeles aberrare = kõrvale kalduma) on mõnevõrra laiem ja hõlmab kõiki muutusi, mida on võimalik valgusmikroskoobiga tuvastada.
Mutatsioonidel võib olla väga erinev mõju:
- Vaikivad mutatsioonid, s.o mutatsioonid, mille korral muutusel ei ole mõju isendile ega nende järglastele, on kromosoomide kõrvalekallete suhtes pigem ebatüüpilised ja neid leidub sagedamini geeni- või punktmutatsioonide piirkonnas.
- Räägitakse funktsiooni kadumise mutatsioonist, kui mutatsiooni tulemusel tekib valesti kokku pandud ja seetõttu funktsioneerimatu valk või puudub see üldse.
- Nn funktsiooni suurendamise mutatsioonid muudavad toime tüüpi või toodetud valkude kogust nii, et tekivad täiesti uued mõjud. Ühelt poolt on see evolutsiooni ja seeläbi liigi püsimajäämise või uute liikide tekkimise jaoks ülioluline mehhanism, kuid teisest küljest, nagu Philadelphia kromosoomi puhul, võib see ka otsustavalt kaasa aidata vähirakkude areng.
Kromosoomide kõrvalekallete erinevatest vormidest on kõige tuntumad arvulised kõrvalekalded, milles üksikud kromosoomid esinevad ainult üks kord (monosoomia) või isegi kolmekordsed (trisoomia).
Kui see kehtib ainult ühe kromosoomi kohta, nimetatakse seda aneuploidiaks ja kogu kromosoomikomplekti mõjutab polüploidia (tri- ja tetraploidsus). Enamasti tekib see valesti jaotumine sugurakkude arengus kromosoomide lahutamise (mittesajustumise) kaudu rakkude jagunemisel (meioos). See viib kromosoomide ebaühtlase jaotumiseni tütarrakkudes ja seega lapse arvulise aberratsioonini.
Mittesuguliste kromosoomide (= autosoomid) monosoomiad ei ühildu eluga ja seetõttu ei esine neid elus lastel. Välja arvatud trisoomiad 13, 18 ja 21, viivad autosoomsed trisoomiad peaaegu alati spontaansete abortideni.
Igal juhul on vastupidiselt sugukromosoomide kõrvalekalletele, mis võivad samuti olla silmapaistmatud, alati tõsiseid kliinilisi sümptomeid ja reeglina enam-vähem väljendunud väliseid kõrvalekaldeid (düsmorfismid).
Selline valesti jaotumine võib toimuda ka hilisemas elus koos mitootiliste rakkude jagunemisega (kõik rakud, välja arvatud sugurakud). Kuna lisaks mõjutatud rakkudele on muutmata rakke, räägitakse somaatilisest mosaiigist. Somaatiliste (kreeka soma = keha) all mõeldakse kõiki rakke, mis ei ole sugurakud. Kuna see mõjutab ainult väikest osa keha rakkudest, on sümptomid tavaliselt palju kergemad. Seetõttu jäävad mosaiigitüübid sageli pikka aega avastamata.
Siit saate teada kõike teema kohta: Kromosoomimutatsioon
Mis on kromosomaalne aberratsioon?
Struktuurne kromosoomide kõrvalekalle vastab põhimõtteliselt kromosoomimutatsiooni määratlusele (vt eespool). Kui geneetilise materjali kogus jääb samaks ja jaotub lihtsalt erinevalt, räägitakse tasakaalustatud aberratsioonist.
Seda tehakse sageli translokatsiooni teel, see tähendab kromosoomisegmendi ülekandmine teise kromosoomi. Kui see on kahe kromosoomi vahetus, siis räägitakse vastastikusest translokatsioonist. Kuna valkude tootmiseks on vaja ainult umbes 2% genoomist, on tõenäosus, et selline geen on murdepunktis ja seeläbi oma funktsiooni kaotanud või selles häiritud, väga väike. Seetõttu jääb selline tasakaalustatud kõrvalekalle sageli märkamata ja kandub edasi mitme põlvkonna jooksul.
See võib aga põhjustada sugurakkude arengus kromosoomide valesti jaotumist, mis võib põhjustada viljatust, spontaanseid raseduse katkemisi või tasakaalustamata kõrvalekalletega järglasi.
Tasakaalustamata kõrvalekalle võib esineda ka spontaanselt, st ilma perekonna ajaloota. Tõenäosus, et laps sünnib elus tasakaalustamata aberratsiooniga, sõltub suuresti mõjutatud kromosoomidest ja varieerub vahemikus 0–60%. See toob kaasa kromosoomisegmendi kadumise (= kustutamine) või dubleerimise (= dubleerimise). Selles kontekstis räägitakse ka osalistest mono- ja trisoomiatest.
Mõnel juhul esinevad need koos kahes erinevas piirkonnas, kusjuures osaline monosoomia on tavaliselt kliiniliste sümptomite ilmnemisel määravam. Need on silmapaistvad näited kustutamisest Kassi karjumise sündroom ja Wolf-Hirschhorni sündroom.
Räägitakse mikrodeletsioonist, kui muutust ei saa enam valgusmikroskoobiga kindlaks teha, s.t kui tegemist on ühe või mõne geeni kadumisega. Seda nähtust peetakse Prader-Willi sündroomi ja Angelmani sündroomi põhjuseks ning see on tihedalt seotud retionoblastoomi arenguga.
Robertsoni ümberasustamine on erijuhtum:
Kaks akrotsentrilist kromosoomi (13, 14, 15, 21, 22) ühinevad oma tsentromeeril ja moodustavad ühe kromosoomi pärast seda, kui lühikesed käed on kadunud (vt struktuur). Kuigi selle tulemuseks on kromosoomide arvu vähenemine, nimetatakse seda tasakaalustatud aberratsiooniks, kuna nende kromosoomide lühikeste käte kadu saab hõlpsasti kompenseerida. Ka siin on mõjud sageli märgatavad alles järgmistes põlvkondades, kuna raseduse katkemise või trisoomiaga elavate laste tõenäosus on väga suur.
Kui kromosoomis on kaks purunemist, võib juhtuda, et vahesegment pööratakse 180 ° ja lülitub kromosoomi. See protsess, mida nimetatakse inversiooniks, on tasakaalust väljas vaid siis, kui murdepunkt asub aktiivses geenis (2% kogu geneetilisest materjalist). Sõltuvalt sellest, kas tsentromeer asub tagurpidi segmendi sees või väljaspool, on see peri- või paratsentriline inversioon. Need muutused võivad aidata kaasa ka geneetilise materjali ebaühtlasele jaotumisele sugurakkudele.
Paratsentrilise inversiooni korral, kui tsentromeer ei ole tagurpidi segmendis, võivad ilmneda ka kahe või ilma tsentromeerita sugurakud. Selle tagajärjel kaob vastav kromosoom kõige esimeste rakujagunemiste käigus, mis viib peaaegu kindlasti raseduse katkemiseni.
Sisestamine on kromosoomifragmendi paigaldamine mujale. Ka siin mõjutatakse järglasi peamiselt sarnaselt. Rõngaskromosoom võib tekkida eelkõige pärast otsaosade kustutamist. Järjestuste tüüp ja suurus on sümptomite raskuse määravaks. Lisaks võib see põhjustada valesid jaotusi ja seeläbi mosaiigitüüpe keharakkudes.
Kui metafaasiline kromosoom eraldub rakkude jagunemise ajal valesti, võivad tekkida isokromosoomid. Need on kaks täpselt sama kromosoomi, mis koosnevad ainult pikkadest või ainult lühikestest kätest. X-kromosoomi korral võib see avalduda Ulrich-Turneri sündroomina (monosoomia X).
Lisateave selle teema kohta: Kromosomaalne kõrvalekalle
Trisoomia 21
Trisoomia 21, paremini tuntud kui Downi sündroom, on tõenäoliselt kõige levinum arvuline kromosoomide kõrvalekalle elussündinute seas, kusjuures isaseid mõjutab see veidi sagedamini (1,3: 1).
Trisoomia 21 esinemise tõenäosus sõltub erinevatest demograafilistest teguritest, näiteks emade keskmine vanus sünnihetkel, ja varieerub piirkonniti veidi.
95% trisoomiast 21 tekib meioosi (sugurakkude jagunemine) kontekstis jagunemisvea tõttu, nimelt mittejaotus, s.t õe kromatiidide eraldamata jätmine.
Neid tuntakse vabade trisoomiatena ja neid tekib emal 90%, isal 5% ja embrüonaalses genoomis veel 5%.
Veel 3% tuleneb tasakaalustamata translokatsioonidest kas 14. kromosoomis või 21. kromosoomis; 21 translokatsioon, luues normaalse ja topeltkromosoomi 21. Ülejäänud 2% on mosaiigitüübid, kus trisoomiat ei tekkinud sugurakkudes ja seetõttu ei mõjuta see kõiki keharakke. Mosaiigitüübid on sageli nii leebed, et võivad pikka aega täiesti avastamata jääda.
Igal juhul tuleks läbi viia kromosoomi uuring, et eristada sümptomaatiliselt identset vaba trisoomiat võimalikust pärilikust translokatsiooni trisoomiast. Seejärel võib järgneda eelmiste põlvkondade perekonna ajalugu.
Kas see teema huvitab teid? Selle kohta lugege järgmist artiklit: Trisoomia 21
Trisoomia 13
Trisoomia 13 või Patau sündroomi sagedus on 1: 5000 ja see on palju haruldasem kui Downi sündroom. Põhjused (vabad trisoomiad, translokatsioonid ja mosaiigitüübid) ja nende protsentuaalne jaotus on suures osas identsed.
Teoreetiliselt sai ultraheli või PAPP-A testi abil peaaegu kõiki juhtumeid diagnoosida prenataalselt. Kuna PAPP-A test ei ole tingimata tavapäraste uuringute osa, diagnoositakse Kesk-Euroopas umbes 80% juhtudest enne sündi.
Ultrahelis on juba näha kasvujääke, kahepoolset huule- ja suulaelõhet ning ebatavaliselt väikesi silmi (mikroftalmia). Lisaks esinevad tavaliselt erineva raskusastmega esiosa ja näo väärarendid (holoprosentsefaalia).
Kui lobar-vormis on ajupoolkerad peaaegu täielikult eraldatud ja tekivad külgmised vatsakesed, siis pool-lobar-vormis eraldatakse sageli ainult aju tagumine osa ja külgmised vatsakesed puuduvad. Kõige raskemas vormis, alobaarses vormis, ajupoolkerasid ei eraldata.
Pool- või alariba kujuga imikud surevad tavaliselt kohe pärast sündi. Ühe kuu möödudes on suremus umbes 50% elusündidest. Kuni 5. eluaastani suureneb 13. trisoomia suremus 90% -ni. Aju väärarengute tõttu jäävad haiged inimesed enamasti kogu elu voodihaigeks ega saa rääkida, mistõttu nad sõltuvad täielikust hooldusest. Lisaks võivad Trismoie 13-l olla ka kaugeleulatuvad füüsilised ilmingud.
Lisateavet selle teema kohta leiate aadressilt: Trisoomia 13 sündimata lapsel
Trisoomia 16
Põhimõtteliselt on trisoomia 16 kõige tavalisem trisoomia (umbes 32% kõigist trisoomiatest), kuid trisoomia 16-ga elavad lapsed on väga haruldased. Üldiselt esinevad elusündid ainult osaliste trisoomiate või mosaiigitüüpidena. Trisoomiate hulgast vastutab see siiski kõige sagedamini surnult sündinud laste eest: 32-st 100-st raseduse katkemisest, mis on tingitud kromosomaalsetest kõrvalekalletest, saab selle trisoomia vormi.
Seetõttu on dokumenteeritud peamiselt sünnieelseid, st sünnieelseid, tuvastatavaid tunnuseid. Tähelepanuväärsed on siin erinevad südamerikked, aeglustunud kasv, üks nabaarteri (muidu topelt) ja kaela suurenenud läbipaistvus, mida seletatakse vedeliku kogunemisega veel täielikult välja arenemata lümfisüsteemi tõttu ja naha suurenenud elastsusega selles piirkonnas. Lisaks ei taandu sageli füsioloogiline nabavääre, s.o soole suure osa ajutine nihe läbi naba väljapoole, mis on tuntud kui omphalocele või nabaväädi purunemine.
Samuti võib ultraheli abil sageli tuvastada ristuvate sõrmedega paindekontraktuuri. Väheste elusate sünnituste korral on märgatav lihaste üldine hüpotensioon, st üldine lihasnõrkus. See viib joomise nõrkuseni ja võib tagada imiku kunstliku toitmise. Sageli esineb ka nelja sõrmega vagu, mis on trisoomiatele nii iseloomulik. Ka siin on trisoomia esinemissagedus otseselt seotud ema vanusega.
Trisoomia 18
Edwardsi sündroom, s.o trisoomia 18, esineb sagedusega 1: 3000. Sünnieelse diagnostika puhul on see sama mis Patau sündroomi puhul: ka siin võimaldaksid samad uuringud kõik patsiendid enne sündi täielikult üles leida. Põhjusi ja nende jaotust saab võrrelda teiste trisoomiatega (vt trisoomia 21).
Lisaks ilmnevad trisoomias 18 osalised trisoomiad, mis, nagu mosaiigitüübid, viivad kliiniliste kulgideni palju kergemini. Sellega seotud düsmorfismid on äärmiselt iseloomulikud ka Edwardsi sündroomile: sündides on patsientide kehakaal oluliselt vähenenud 2 kg (normaalne: 2,8–4,2 kg), taanduv lai otsmik, üldiselt vähearenenud näo alaosa väikese suuga avanevad, kitsad silmalau pilud ja pööratud tahapoole, kuju muutnud kõrvad (fauni kõrv). Samuti on märgatav pea tagaosa, mis on vastsündinu jaoks ebatavaliselt tugevalt arenenud. Ribid on ebatavaliselt kitsad ja habras. Vastsündinutel on ka kogu lihase püsiv pinge (toon), mis aga pärast esimest paari nädalat ellujäänutel taandub.
Teine iseloomulik tunnus on 2. ja 5. sõrme ristumine üle 3. ja 4. sõrme kokku löövate sõrmede arvuga, samal ajal kui jalad on ebatavaliselt pikad (möödunud), eriti selgelt väljendunud kand, kidurad varbaküüned ja tagumine suur varvas. .
Elundi tõsised väärarengud on tavalised ja esinevad tavaliselt koos: südame- ja neerupuudulikkus, soole väärareng (malrotatsioon), kõhukelme (mesenterium commune) adhesioonid, söögitoru oklusioon (söögitoru atresia) ja palju muud.
Nende väärarengute tõttu on esimese nelja päeva jooksul suremus umbes 50%, üle 5 aasta vanuseks elab vaid umbes 5–10%. Täiskasvanuikka elamine on absoluutne erand.Igal juhul on intellektipuude väga väljendunud ega oska rääkida, on voodihaige ja pidamatus, sõltudes seega täielikult välisest abist.
Trisoomia 18 kohta üksikasjalikuma teabe saamiseks lugege palun ka meie selle teema üksikasjalikku artiklit:
- Trisoomia 18 (Edwardsi sündroom)
- Trisoomia 18 sündimata lapsel
Trisoomia X
Trisoomia X on arvulise kromosomaalse aberratsiooni kõige silmapaistmatum vorm, mõjutatud isikute välimus, kes on loogiliselt kõik naised, ei erine teistest naistest eriti. Mõni paistab silma selle poolest, et on eriti pikk ja mõnevõrra “lihav” näojoonega. Ka vaimne areng võib olla suures osas normaalne, ulatudes normaalsest piirist kuni kerge vaimse puudeni.
Kuid see luure puudujääk on mõnevõrra tõsisem kui teiste sugukromosoomide trisoomiate (XXY ja XYY) puhul. Sagedusega 1: 1000 pole see tegelikult nii haruldane, kuid kuna trisoomiat ei seostata tavaliselt kliiniliselt oluliste sümptomitega, ei diagnoositakse tõenäoliselt enamikul seda haigust põdevatel naistel kogu elu.
Kandjad avastatakse enamasti juhuslikult perekontrolli või sünnieelse diagnostika käigus.Viljakust saab veidi vähendada ja järgmise põlvkonna sugukromosoomide kõrvalekallete määr võib veidi suureneda, nii et kui soovite lapsi saada, soovitatakse geneetilist nõustamist.
Nagu teiste trisoomiate puhul, areneb trisoomia X kõige sagedamini vaba trisoomiana, s.t õekromatiidide jagunemise (mittesajustumise) puudumise tõttu. Ka siin tekib see tavaliselt ema munarakkude küpsemise ajal, kuigi tõenäosus suureneb vanusega.
Habras X sündroom
Meestel eelistatakse habras X sündroomi või Martin Belli sündroomi, kuna neil on ainult üks X kromosoom ja seetõttu on muutus neid rohkem mõjutanud.
See juhtub ühe aasta jooksul elusate meessoost sündide seas sagedusega 1: 1250, mistõttu on see kõige tavalisem mittespetsiifilise vaimse alaarengu vorm, s.t kõik vaimsed puuded, mida ei saa kirjeldada tüüpiliste tunnustega erilise sündroomiga.
Habras X sündroom võib tüdrukutel esineda tavaliselt mõnevõrra nõrgemas vormis, mis on tingitud ühe X kromosoomi juhuslikust inaktiveerimisest. Mida suurem on välja lülitatud terve X-kromosoomi osakaal, seda tugevamad on sümptomid.
Enamasti on naised siiski premutatsiooni kandjad, mis veel kliinilisi sümptomeid ei tekita, kuid suurendab nende poegade täieliku mutatsiooni tõenäosust. Väga harvadel juhtudel võivad mehed olla ka premutatsiooni kandjad, mida nad saavad seejärel edasi anda ainult tütardele, kes on siiski tavaliselt kliiniliselt terved (Shermani paradoks).
Sündroomi põhjustab äärmiselt suurenenud arv CGG kolmikuid (teatud alusjärjestus) FMR geenis (habras sait-vaimne alaareng); 10-50 koopia asemel on eelautatsioon 50-200, kui see on täielikult välja töötatud 200- 2000 eksemplari.
Valgusmikroskoobi all näeb see välja nagu pika käe katkemine, mis andis sündroomile selle nime. See viib mõjutatud geeni deaktiveerimiseni, mis omakorda põhjustab sümptomeid.
Mõjutatud inimesed näitavad kõne ja liikumise aeglustumist ning võivad näidata käitumisprobleeme, mis võivad minna hüperaktiivsuse, aga ka autismi suunas. Puhtalt välised kõrvalekalded (düsmorfismi tunnused) on pikk nägu, millel on silmatorkav lõug ja väljaulatuvad kõrvad. Puberteedieas on munandid sageli oluliselt suurenenud (makroorhiidiad) ja näojooned muutuvad jämedamaks. Premutatsiooni naissoost kandjate hulgas on kerge psühholoogiliste kõrvalekallete kuhjumine ja eriti varajane menopaus.
Mis on kromosoomianalüüs?
Kromosoomianalüüs on tsütogeneetika protsess, mille abil saab tuvastada arvulisi või struktuurseid kromosoomide kõrvalekaldeid.
Sellist analüüsi kasutatakse näiteks juhul, kui viivitamatult kahtlustatakse kromosomaalset sündroomi, st väärarengute (düsmorfismid) või vaimupuude (alaareng), aga ka viljatuse, regulaarsete raseduse katkemiste (abordid) ja ka koos teatud vähid (nt lümfoomid või leukeemia).
Selleks on tavaliselt vaja lümfotsüüte - erilist tüüpi immuunrakke, mis saadakse patsiendi verest. Kuna sel viisil on võimalik saada ainult suhteliselt väike kogus, stimuleeritakse rakke jagunema fütohemaglutiniiniga ja lümfotsüüte saab seejärel kasvatada laboris.
Mõnel juhul võetakse proovid (biopsiad) hoopis nahalt või seljaajult sarnase protseduuriga. Eesmärk on saada võimalikult palju DNA-materjali, mis on praegu rakkude jagunemise keskel. Metafaasis on kõik kromosoomid paigutatud ühele tasandile ligikaudu raku keskele, et neid järgmises etapis, anafaasis, tõmmata raku vastaskülgedele (poolustele).
Sel hetkel on kromosoomid eriti tihedalt (väga kondenseerunud). Lisatakse spindlimürk kolhitsiin, mis töötab täpselt selles rakutsükli faasis, nii et metafaasilised kromosoomid kogunevad. Seejärel isoleeritakse ja värvitakse spetsiaalsete värvimismeetodite abil.
Kõige tavalisem on GTG sidumine, mille käigus kromosoome töödeldakse trüpsiini, seedeensüümi ja pigmendiga Giemsa. Eriti tihedalt asustatud piirkonnad ning adeniini- ja tümiinirikkad piirkonnad on tumedad.
Saadud G-ribad on iseloomulikud igale kromosoomile ja neid peetakse lihtsustatult vähemate geenidega piirkondadeks. Sel viisil värvitud kromosoomidest tehakse pilt tuhandekordsel suurendusel ja arvutiprogrammi abil luuakse karüogramm. Lisaks ribamustrile kasutatakse kromosoomide suuruse ja tsentromere asukohta, et aidata kromosoome vastavalt korraldada. Kuid on ka muid ribamismeetodeid, millel võivad olla väga erinevad eelised.
Toimetuse soovitused
Üldist teavet leiate järgmistest artiklitest:
- Rakutuuma jagunemine
- Rakutuuma funktsioonid
- Trisoomia 21
- Geneetilised haigused